
深度揭秘“合成致死”药物PARP抑制剂杀死癌细胞的机制
小秋走了快两年了, 小秋妈妈在深夜两点看了我们发的肿瘤治疗进展的帖子,发来寄语, “ 医学研究水平一直都在进步, 直到彻底消灭癌症这个大坏蛋….”
“欲使其灭亡,必先使其疯狂。”
这句话用在癌细胞身上,恐怕再适合不过了。
从本质上讲,癌症就是一种基因病。当细胞内的基因突变积累到一定程度之后,细胞要么走向衰老死亡,要么就走向癌变。
不过这些突变在赋予癌细胞不死和无限繁殖能力的同时,也给它们的毁灭埋下了伏笔。
这个毁灭伏笔的序曲在1922年。
那一年,在哥伦比亚大学摩尔根实验室工作的遗传学家Calvin Bridges,在黑腹果蝇身上发现一种有趣的现象:当某两个特定的基因同时突变失活时,会导致果蝇的死亡;而这两个基因单独任何一个突变失活,都不会给果蝇带来致命的伤害[1]。
1946年,Theodosius Dobzhansky给这种现象取了个名字,它就是今天大名鼎鼎的“合成致死”效应[2]。
这个概念一沉寂就是51年。在考虑到癌细胞携带有大量基因突变之后,1997年,福瑞德·哈金森癌症研究中心的Stephen Friend敏锐地察觉到,这个“合成致死”的理念或许可以用到癌症的治疗中[3]。
在Stephen Friend看来,正常细胞癌变是个异常的举动。俗话说,“物极必反”,那我们干脆就让异常来的更疯狂吧。
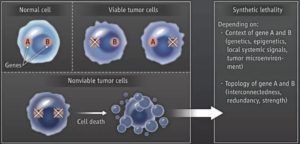
合成致死理念
这个想法很疯狂,不过竟真的能让癌细胞走向死亡。
2014年,全世界第一个按照“合成致死”理念设计的抗癌药物PARP(多聚ADP核糖聚合酶)抑制剂Olaparib,获得FDA批准用于治疗卵巢癌。随后,在2016年和2017年,PARP抑制剂Rucaparib和Niraparib先后闪亮登场。
一种全新的抗癌手段崛地而起。
“破罐子破摔”
实际上,大部分细胞从正常走向癌变,并不是说它们的基因天生就不好,而是因为在生长的过程中,细胞的DNA会不断遭受内在和周遭各种不利因素的夹击,例如,辐射、化学毒物、细胞自身有害代谢产物、DNA自己复制错误等,导致癌症相关基因发生了突变,最终导致了癌症[4]。
据估计,人体每个细胞每天产生的单链DNA损伤数约为10000个,如果把其他损伤也都算上的话这个数据又要翻10倍,变成10万个[5]。
与DNA遭受的损伤相比,癌症的发生风险就显得微不足道了,这主要得益于人体精密、复杂而高效的DNA修复系统。
在DNA损伤中,最严重的损伤是单链断裂和双链断裂,不过单链断裂更常见。这些断裂如果不能得到及时、准确的修复,会使基因组变得不稳定,进而引起癌变,甚至直接导致细胞死亡。

为维持正常生理功能,细胞必须有多种DNA损伤发现和修复机制,使受损的DNA得到及时精确的修复。
对于单链断裂而言,它的修复主要依赖于PARP,这个酶在人体内有17种,它们虽然长得有些像,但功能却不尽相同[6]。
目前的研究认为,DNA损伤修复依赖的PARPs主要包括PARP-1和PARP-2,它俩都能精准地识别DNA的伤口,并与DNA亲密结合。只不过在修复DNA损伤的过程中,PARP-1发挥着90%以上的功能,PARP-2更像是个备胎[7]。
而对于双链断裂而言,它虽然少,但是情况更严重,如果不能及时修复,细胞的DNA就会变得不稳定,细胞最终走向死亡。
所以双链DNA断裂有两种主要的修复方式。一种是非同源末端连接(NHEJ)修复,它更像个紧急救火队长,先不管修复的对不对,把断掉的DNA连上再说。这种方法最主要的优点是快,但是非常容易出错,一旦出大问题,对细胞来说有可能就是毁灭性的打击。
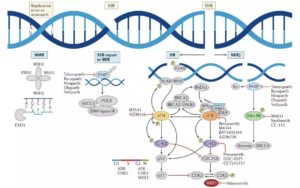
另外一种是同源重组(HR)修复途径,参与这种修复方式的蛋白非常之多例如BRCA、ATM、RAD51等等,其中最为人所熟知的是BRCA蛋白。这种修复方式像外科手术,是一种高保真、无错误的修复方式。
对于癌细胞而言,既然它是基因突变导致的,那肯定是上述修复过程没起作用,或者工作不到位造成的。
鉴于癌细胞也要维持自身基因组的稳定性,因此,作为一个“理性”的癌细胞,它们肯定不会让上述所有的DNA损伤修复机制全部瘫痪。不过为了保持进化的活力,部分修复方式失去功能是可能的。
这也就给了科学家们可乘之机。以DNA修复为靶点,把癌细胞这个DNA已经出现大量突变的“破罐子”彻底捣毁。
2005年,“摔破”癌细胞这个“破罐子”的曙光初现。
两个独立研究团队背靠背在顶级期刊《自然》发表重要研究成果,首次证实PARP抑制剂与BRCA1或BRCA2突变之间存在“合成致死”的相互作用[8,9]。
合成致死治疗癌症的大门打开了。
“扼住”PARP的咽喉
结合前面介绍的DNA修复机制,你会发现PARP与BRCA是一对合成致死冤家这事儿并不难理解。癌细胞的DNA再混乱,它们也还是需要维持自身基因组的稳定。
如果负责双链断裂修复的BRCA突变失活了,我们再把管单链断裂的PARP抑制掉,癌细胞中每天出现的大量单链断裂就会变成双链断裂,最终导致癌细胞死亡。
不过,这个合成致死的机制看似简单,其实要设计一个优秀的PARP抑制剂并没有那么简单。
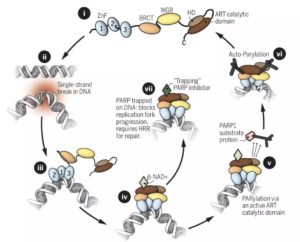
PARP抑制剂“合成致死”机制
要把这个事情说清楚,我们还得从PARP修复单链DNA断裂的过程说起。
在细胞内,一旦PARP发现DNA上存在单链断裂的缺口,就会立即结合上去,这种结合会激活PARP的催化活性。
此时,游荡在PARP周围的烟酰胺腺嘌呤二核苷酸(NAD+,这个物质最近非常火,抗衰老、抗癌都有它的份儿)会立即与PARP的活性位点结合,结合后的复合体会把周围参与DNA修复效应子统统拉过来,填补上DNA断开的缺口。于此同时,染色质也会变得松弛,PARP复合体就顺利从损伤缺口脱离下来,回到之前的失活状态待命[10]。
在这个修复的过程中,NAD+与PARP的结合,就是那个关键的点。
实际上,早在30年前,小分子烟酰胺类似物就被证明可以竞争性抑制这个过程,并增强DNA损伤剂硫酸二甲酯的细胞毒性[11-13]。
目前在临床中使用的所有PARP抑制剂,都有一个与NAD+竞争结合PARP的烟酰胺部分,因此它们抑制PARP催化活性的能力是类似的;然而,由于不同的抑制剂结构存在较大差异,它们对不同PARP家族成员的选择性存在一定的差异[14]。
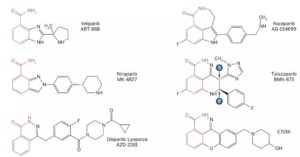
PARP抑制剂结构比拼:红色部分为共通部分
就拿已经获得FDA批准的三个PARP抑制剂而言,有研究表明,与Olaparib和Rucaparib相比,Niraparib只选择性抑制PARP-1和PARP-2的活性,不抑制PARP-3的活性[15,16]。
可别小看了这个差异,毕竟科学家对PARP-3的认知还不够。
虽然从结构上看,PARP-3与PARP-1也长得很像,但PARP-3在组织分布、生物学功能方面,与PARP-1却表现出很大的不同[17]。
除此之外,之前还有研究表明,当PARP-1表达被抑制之后,PARP-2的表达会代偿性地增加,以顶替PARP-1的职能;但PARP-3却不会在PARP-1和PARP-2表达被抑制后代偿性增加[17-19]。
而且也有研究表明,特异性抑制PARP-1和PARP-2,但不抑制PARP-3,就能让肿瘤消退[20]。
这些似乎都表明PARP-3有其独特的生物学功能。
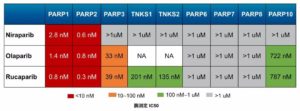
部分PARP抑制剂选择性的比较
由此可见,PARP抑制剂抑制PARP-3的活性,可能不仅没有抗癌效果,而且可能还有意想不到的副作用。
“诱捕”PARP
在研究PARP抑制剂的过程中,科学家们还发现了一个很奇怪的现象。
PARP抑制剂对癌细胞的杀伤力大于敲除PARP基因本身[21,22],这意味着PARP抑制剂的抗癌效果不仅仅在于抑制PARP的活性,背后可能还有其他的原因。
后来科学家发现,这个现象要归结于PARP抑制剂对PARP的“诱捕”作用[21,22]。
所谓“诱捕”作用,说的是PARP抑制剂竞争性结合到PARP酶上之后,会导致与受损DNA结合的PARP-1和PARP-2被困在DNA上下不来了,同时直接造成其他的DNA修复蛋白也结合不上来了。后果是,DNA断裂不仅不能被修复,而且还从单链断裂变成双链断裂,最终导致细胞死亡[23]。
实际上,科学家已经认识到,“诱捕”PARP并把它“钉”在DNA上,才是PARP抑制剂消灭癌细胞的最大杀器[18,21]。因此,在比较单一PARP抑制剂抗癌活性时,必须基于其捕获效力。
从这个角度看,目前已经获FDA批准的三款PARP抑制剂捕获PARP的能力依次为:Niraparib > Olaparib = Rucaparib[21,22,24]。
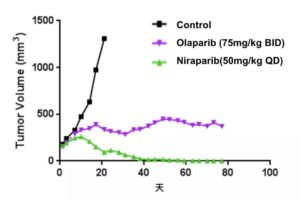
PARP抑制剂在BRCA2突变模式小鼠中的效果
它们捕获能力之间的这种差异,也反映在了抗癌效果上。
与众不同,潜力无限
可能你已经发现了,上面介绍的都是PARP抑制剂与BRCA突变之间的协同致死作用,但是以BRCA为代表的同源重组通路并非只有BRCA这一条路,对于BRCA基因没有突变的癌细胞,PARP抑制剂是不是也有效果呢?
其实,2005年的研究就已经表明:PARP抑制剂对于BRCA没有突变的癌细胞也有杀伤力。
只不过与携带BRCA突变的癌细胞相比,BRCA没有突变的癌细胞对PARP抑制剂的敏感性差了近1000倍[9]。
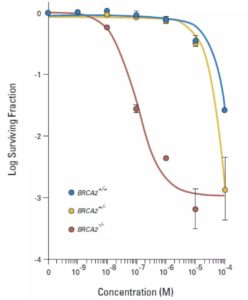
BRCA突变比不突变对PARP抑制剂更敏感
这也就意味着,对于那些BRCA基因没有突变的肿瘤,在使用PARP抑制剂治疗时,需要更高的药物暴露,才能到达与BRCA突变的肿瘤同样的效果。
对于这种情况而言,哪种PARP抑制剂的效果好,就要看每种药物药物在体内吸收、分布、代谢和排泄规律的数据。
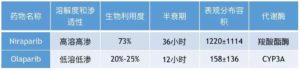
从上面的表格不难看出,Niraparib的药代学数据有一定的优势,它的高溶解和渗透性,让它有个非常高的生物利用度,这会让它更容易抵达肿瘤;长达36个小时的半衰期,大大降低了给药的频率,一天一次肯定比一天两次更方便。
此外,CYP3A是个被盯上概率比较高的靶点,目前有不少的药物的作用就是抑制它的活性,还有一些药物会诱导CYP3A的表达,这都会影响以CYP3A为代谢酶的药物的使用;而以羧酸酯酶为代谢酶的Niraparib基本不需要考虑药物之间的相互作用。
再加上其在PARP酶捕获上的优势。使得Niraparib不仅在BRCA野生型/HRD阳性的卵巢癌PDX模型中显示出更强的抗肿瘤活性;而且在BRCA野生型/HRD阴性的卵巢癌PDX模型中也表现出更强的抗肿瘤活性[25]。
正是基于以上优秀的临床前数据,以及后来开展的临床研究。在2017年,Niraparib获得FDA的上市批准,成为全球第一个适用于所有铂敏感、复发卵巢癌患者,无论其BRCA基因是否突变的PARP抑制剂。
这也就意味着,在使用Niraparib之前不需进行BRCA或其它生物标志物检测,这显然能让更多卵巢癌患者获益。
当然,我们对于PARP抑制剂的认知还刚刚起步,它应该还有很多未知的技能等着我们去发现。
例如最近德州大学MD安德森癌症中心的研究团队发现,抑制肿瘤细胞的PARP修复通路,竟然可以触发STING免疫通路,进而募集杀伤性T细胞进入肿瘤[26]。
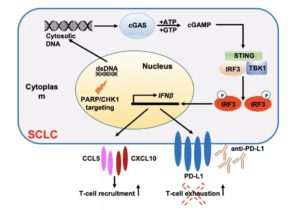
抑制PARP激活免疫通路的机制
这个研究暗示,PARP抑制剂联合免疫检查点抑制剂将大有可为。
PARP抑制剂的未来,可期。
参考资料:
[1]:Nijman S M, Friend S H. Potential of the Synthetic Lethality Principle[J]. Science, 2013, 342(6160): 809-811. DOI:10.1126/science.1244669
[2].Dobzhansky T. GENETICS OF NATURAL POPULATIONS. XIII. RECOMBINATION AND VARIABILITY IN POPULATIONS OF DROSOPHILA PSEUDOOBSCURA[J]. Genetics, 1946, 31(3): 269-290.
[3].Hartwell L, Szankasi P, Roberts C J, et al. Integrating Genetic Approaches into the Discovery of Anticancer Drugs[J]. Science, 1997, 278(5340): 1064-1068. DOI:10.1126/science.278.5340.1064
[4].Pilie P G, Tang C, Mills G B, et al. State-of-the-art strategies for targeting the DNA damage response in cancer[J]. Nature Reviews Clinical Oncology, 2018. DOI:10.1038/s41571-018-0114-z
[5].Hoeijmakers J H. DNA Damage, Aging, and Cancer[J]. The New England Journal of Medicine, 2009, 361(15): 1475-1485. DOI:10.1056/NEJMra0804615
[6].Peraltaleal A, Rodriguezvargas J M, Aguilarquesada R, et al. PARP inhibitors: New partners in the therapy of cancer and inflammatory diseases[J]. Free Radical Biology and Medicine, 2009, 47(1): 13-26. DOI:10.1016/j.freeradbiomed.2009.04.008
[7].Langelier M, Riccio A A, Pascal J M, et al. PARP-2 and PARP-3 are selectively activated by 5′ phosphorylated DNA breaks through an allosteric regulatory mechanism shared with PARP-1[J]. Nucleic Acids Research, 2014, 42(12): 7762-7775. DOI:10.1093/nar/gku474
[8].Bryant H E, Schultz N, Thomas H D, et al. Specific killing of BRCA2-deficient tumours with inhibitors of poly(ADP-ribose) polymerase[J]. Nature, 2005, 434(7035): 913-917. DOI:10.1038/nature03443
[9].Farmer H, Mccabe N, Lord C J, et al. Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy.[J]. Nature, 2005, 434(7035): 917-921. DOI:10.1038/nature03445
[10].Lord C J, Ashworth A. PARP inhibitors: Synthetic lethality in the clinic[J]. Science, 2017, 355(6330): 1152-1158. DOI:10.1126/science.aam7344
[11].Miwa M, Tanaka M, Shinshi H, et al. Seminar on Poly(ADP-Ribose) and ADP-ribosylation of Protine[J]. Journal of Biochemistry, 1975: 10-11. DOI:10.1093/oxfordjournals.jbchem.a130855
[12].Purnell M R, Whish W J. Novel inhibitors of poly(ADP-ribose) synthetase.[J]. Biochemical Journal, 1980, 185(3): 775-777. DOI:10.1042/bj1850775
[13].Terada M, Fujiki H, Marks P A, et al. Induction of erythroid differentiation of murine erythroleukemia cells by nicotinamide and related compounds[J]. Proceedings of the National Academy of Sciences of the United States of America, 1979, 76(12): 6411-6414. DOI:10.1073/pnas.76.12.6411
[14].Pilie P G, Tang C, Mills G B, et al. State-of-the-art strategies for targeting the DNA damage response in cancer[J]. Nature Reviews Clinical Oncology, 2018. DOI:10.1038/s41571-018-0114-z
[15]. Wang S et al. Presented at AACR-NCI-EORTC Molecular Targets and Cancer Therapeutics Symposium; Nov 29–Dec 2, 2016; Munich, Germany;
[16].Zhong Y, Katavolos P, Nguyen T, et al. Tankyrase Inhibition Causes Reversible Intestinal Toxicity in Mice with a Therapeutic Index < 1[J]. Toxicologic Pathology, 2016, 44(2): 267-278. DOI:10.1177/0192623315621192
[17].Beck C, Robert I, Reinasanmartin B, et al. Poly(ADP-ribose) polymerases in double-strand break repair: focus on PARP1, PARP2 and PARP3.[J]. Experimental Cell Research, 2014, 329(1): 18-25. DOI:10.1016/j.yexcr.2014.07.003
[18].Ghosh R, Roy S, Kamyab J, et al. Common and unique genetic interactions of the poly(ADP-ribose) polymerases PARP1 and PARP2 with DNA double-strand break repair pathways.[J]. DNA Repair, 2016: 56-62. DOI:10.1016/j.dnarep.2016.06.001
[19].Sukhanova M V, Abrakhi S, Joshi V, et al. Single molecule detection of PARP1 and PARP2 interaction with DNA strand breaks and their poly(ADP-ribosyl)ation using high-resolution AFM imaging[J]. Nucleic Acids Research, 2016, 44(6). DOI:10.1093/nar/gkv1476
[20].Oconnor L O, Rulten S L, Cranston A, et al. The PARP Inhibitor AZD2461 Provides Insights into the Role of PARP3 Inhibition for Both Synthetic Lethality and Tolerability with Chemotherapy in Preclinical Models.[J]. Cancer Research, 2016, 76(20): 6084-6094. DOI:10.1158/0008-5472.CAN-15-3240
[21].Murai J, Huang S Y, Das B B, et al. Trapping of PARP1 and PARP2 by Clinical PARP Inhibitors[J]. Cancer Research, 2012, 72(21): 5588-5599. DOI:10.1158/0008-5472.CAN-12-2753
[22].Murai J, Huang S N, Renaud A, et al. Stereospecific PARP trapping by BMN 673 and comparison with olaparib and rucaparib.[J]. Molecular Cancer Therapeutics, 2014, 13(2): 433-443. DOI:10.1158/1535-7163.MCT-13-0803
[23].Satoh M S, Lindahl T. Role of poly(ADP-ribose) formation in DNA repair.[J]. Nature, 1992, 356(6367): 356-358. DOI:10.1038/356356a0
[24].Pommier Y, Oconnor M J, De Bono J S, et al. Laying a trap to kill cancer cells: PARP inhibitors and their mechanisms of action[J]. Science Translational Medicine, 2016, 8(362). DOI:10.1126/scitranslmed.aaf9246
[25].Sun K, Mikule K, Wang Z, et al. A comparative pharmacokinetic study of PARP inhibitors demonstrates favorable properties for niraparib efficacy in preclinical tumor models[J]. Oncotarget, 2018, 9(98). DOI:10.18632/oncotarget.26354
[26].Sen T, Rodriguez B L, Chen L, et al. Targeting DNA damage response promotes anti-tumor immunity through STING-mediated T-cell activation in small cell lung cancer[J]. Cancer Discovery, 2019: CD-18-1020. DOI:10.1158/2159-8290.CD-18-1020
